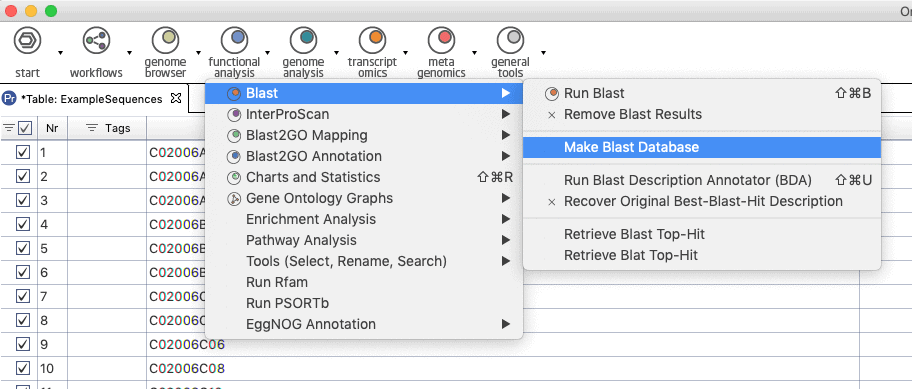
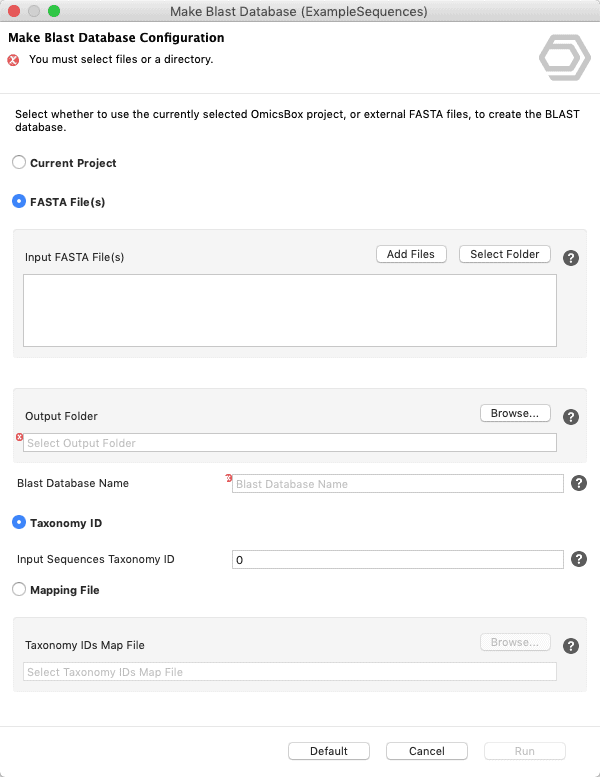
Update: Since Blast2GO v.4 or in OmicsBox you can create your Blast Database directly from within the application. See screenshots below.
Note: This tutorial is based on the NCBI blast binaries released in 2014 and some parameters might have changed since then
If you want to blast your sequences against an own own database you need to create a custom NCBI Blast Database from your FASTA file. If you intent to use the results later on in Blast2GO or OmicsBox for functional annotation etc. you have to be careful with the formatting because Blast2GO/OmicsBox will need the accession IDs information in order to execute the Gene Ontology mapping for the functional annotation step.
This tutorial will guide you on how to format your own database from a FASTA file with the command line and how to use the correct parameters to run the a local blast. Note: This can also be done from within OmicsBox without the command line.
General:
- The sequences have to be in FASTA format and the accession IDs in between “|”.
>ref|AccID|sequence definition KLPPGILVSDKAIKENEESSLLRDTHMISMTRKITDKL KSGFSSFFTLFSRKLIRTTLLLWVLFFANAFSYYGAVLLTSKLSSGDSKCGSKVLHADKS KDNSLYVDVFITSFAELPGLILSAIIVDKIGRKLSMVLMFVLACIFLLPLVFHQSAVVTTVL LFGVRMCATGTITVATIYAPEIYPTSARTTGAGVASAVGR
- Download and use the Blast+ executables (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/) from NCBI to format the database.
- When formatting the database do not forget to use the “parse the sequence IDs” parameter, because they are needed for the Gene Ontology mapping step in Blast2GO or OmicsBox.
./makeblastdb -dbtype prot -in /path/to/yourfilewithproteinsequence.fasta -parse_seqids -out myformattedDBname
- Now you can blast your sequences against the formatted database, either using the Local Blast in OmicsBox or via command line.
./blastx -db ~/path/to/your/myformattedDBname/ -outfmt 5 -evalue 1e-3 -word_size 3 -show_gis -num_alignments 20 -max_hsps 20 -num_threads 5 -out local_blast.xml -query myDNAsequenceTOblast.fasta
Note: When blasting your sequences, make sure you use the parameter -show_gis in order to retrieve the accessions IDs from the formatted database. The resulting file will be an xml file (-outfmt 5), which can be easily loaded in Blast2GO.
- Finally, load your blast xml-file results (local_blast.xml) into Blast2GO (File -> Load -> Load Blast Results -> XML files) and visualize several Blast results (Show Blast Results) to see if the accession appears in the right place (ACC).
Example:
Please have a look at the following example as there is the need to use the “sed” command line in Linux in order to have the fasta file in the desired format.
- Download this fasta Viridiplantae from Uniprot.
- Open a terminal window and see how the fasta file looks like.
head uniprotkb_viridiplantae.fasta >TR:A0A022_9ACTO A0A022 Putative dehydrogenase OS=Streptomyces ghanaensis PE=4 SV=1 MPSMLDAVVVGAGPNGLTAAVELARRGFSVALFEARDTVGGGARTEELTLPGFRHDPCSA
Note: Have a look at the first line of the fasta file. The accession ID A0A022 is not in between “|”. There is the need to reformat the whole fasta for the accessions IDs.
- Rectify the fasta file in order to have the correct format.
sed -E 's/(>[A-Za-z]+):([A-Z0-9]+)_(.*)/\1|\2|\3/g' uniprotkb_viridiplantae.fasta > uniprotkb_viridiplantae_mod.fasta
Note: This is an example and not an universal command. The user will need to understand from their sequences how to change them in order to obtain the correct format.
- Let us have a look at the modified file.
head uniprotkb_viridiplantae_mod.fasta >TR|A0A022|9ACTO A0A022 Putative dehydrogenase OS=Streptomyces ghanaensis PE=4 SV=1 MPSMLDAVVVGAGPNGLTAAVELARRGFSVALFEARDTVGGGARTEELTLPGFRHDPCSA
Now it looks very similar to what we wanted.
- It is now safe to create the database using the Blast+ executables.
./makeblastdb -dbtype prot -in /path/to/uniprotkb_viridiplantae_mod.fasta -parse_seqids -out uniprotkb_viridiplantae_mod_db
- Run blast.
./blastx -db ~/path/to/your/uniprotkb_viridiplantae_mod_db/ -outfmt 5 -evalue 1e-3 -word_size 3 -show_gis -num_alignments 20 -max_hsps 20 -num_threads 5 -out local_blast.xml -query 10_seq.fasta
- Load your local_blast.xml file into Blast2GO (File -> Load -> Load Blast Results -> XML files) and visualize several Blast results (Show Blast Results) to see if the accession appears in the right place (ACC).
- You can proceed with the mapping step as usual.